Core AI/ML Concepts for Bioinformatics
Bioinformatics has always been deeply rooted in computational methods, statistical modeling, and algorithmic design. However, the increasing volume, heterogeneity, and dimensionality of biological data have introduced challenges that often outpace traditional approaches. As a result, AI and ML are becoming central to modern bioinformatics workflows. This foundational module is intended to support bioinformaticians in understanding and adopting AI methods that align with their scientific goals.
From Algorithms to Learning Systems
Traditional bioinformatics relies on manually crafted rules or probabilistic models to solve problems such as sequence alignment, gene annotation, or motif discovery. While effective, these rule-based or statistical models often fall short in capturing the complex, nonlinear relationships present in biological systems. In contrast, machine learning systems learn patterns directly from data, making them more flexible and scalable for high-dimensional tasks.
Machine learning is broadly divided into supervised, unsupervised, and semi-supervised approaches. Supervised learning involves learning a function that maps inputs to known outputs. Examples in bioinformatics include predicting the effect of single nucleotide variants on protein function or classifying cell types from single-cell RNA-seq data. Unsupervised learning, in contrast, infers structure in unlabeled data. Common applications include clustering gene expression profiles or dimensionality reduction for visualization, such as t-SNE and UMAP.
Semi-supervised and transfer learning approaches are particularly valuable in biological domains where annotated data is sparse or expensive to generate. These models allow for the reuse of representations learned from large public datasets and can be fine-tuned for specific biological problems.
Key Methods and Tools
Supervised learning techniques commonly used in bioinformatics include support vector machines (SVMs), random forests, gradient-boosted decision trees, and deep neural networks. Each has been successfully applied across a range of genomics problems. For example, deep neural networks have been used to predict splicing from raw DNA sequences with higher accuracy than previous models (Jaganathan et al., 2019). Similarly, boosted models such as XGBoost are frequently used in feature-rich environments such as cancer classification or variant prioritization (Chen and Guestrin, 2016).
Unsupervised methods play a central role in tasks such as cell clustering in single-cell experiments or identifying latent factors in transcriptomic studies. Dimensionality reduction tools like UMAP have become standard for visualizing complex relationships in scRNA-seq data (Becht et al., 2019).
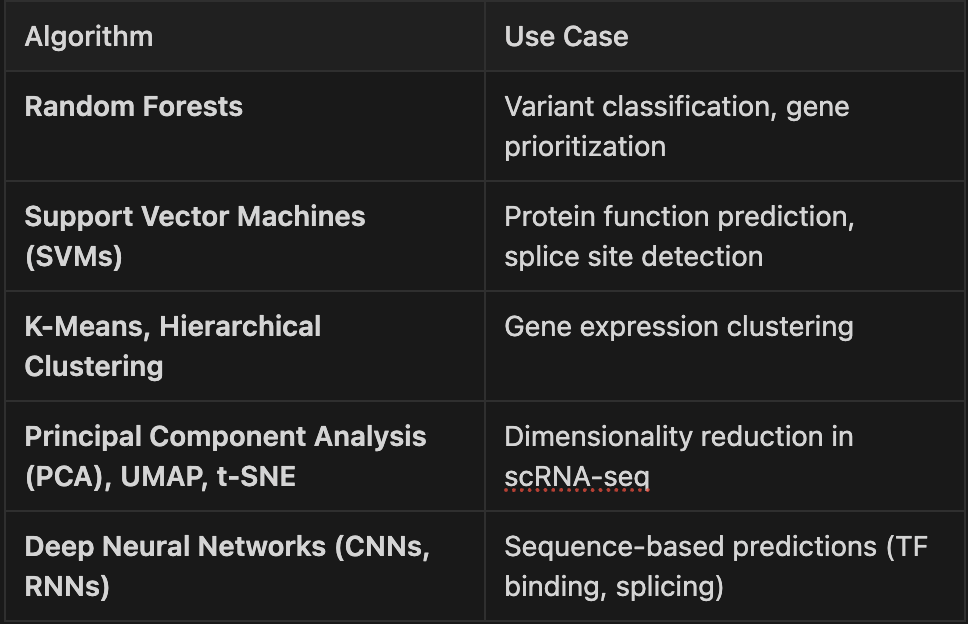
Key methods and tools
Several open-source libraries support the application of these techniques in bioinformatics

Possible options
Model Evaluation and Best Practices
Rigorous model evaluation is essential for robust conclusions. Performance metrics such as accuracy, precision, recall, F1 score, and area under the receiver operating characteristic curve (AUC-ROC) are commonly used, depending on the context of the biological problem. For imbalanced data, the area under the precision-recall curve (AUC-PR) is often more informative.
Cross-validation and external testing on held-out datasets help to prevent overfitting and ensure generalizability. Bioinformaticians should pay close attention to data leakage, sample independence, and appropriate partitioning strategies, particularly when working with high-throughput data from multiple batches or studies.
Bioinformaticians already possess a computational mindset, familiarity with scripting and data preprocessing, and domain knowledge of molecular biology. These assets provide a strong foundation for learning and applying machine learning techniques. As biological datasets grow in size and complexity, ML methods enable the discovery of subtle, nonlinear patterns that would be missed by classical statistical tests.
For instance, in functional genomics, deep learning models have been used to predict protein-DNA and protein-RNA binding preferences directly from sequence, outperforming motif-based approaches . In cancer transcriptomics, variational autoencoders have revealed latent representations associated with molecular subtypes and prognosis. Rather than replacing existing tools, AI augments the bioinformatician’s toolkit.
References
Becht, E., McInnes, L., Healy, J., Dutertre, C. A., Kwok, I. W. H., Ng, L. G., Ginhoux, F., & Newell, E. W. (2019). Dimensionality reduction for visualizing single-cell data using UMAP. Nature Biotechnology, 37, 38–44. https://doi.org/10.1038/nbt.4314
Eraslan, G., Avsec, Ž., Gagneur, J., & Theis, F. J. (2019). Deep learning: New computational modelling techniques for genomics. Nature Reviews Genetics, 20(7), 389–403. https://doi.org/10.1038/s41576-019-0122-6
Golub, T. R., Slonim, D. K., Tamayo, P., Huard, C., Gaasenbeek, M., Mesirov, J. P., Coller, H., Loh, M. L., Downing, J. R., Caligiuri, M. A., Bloomfield, C. D., & Lander, E. S. (1999). Molecular classification of cancer: Class discovery and class prediction by gene expression monitoring. Science, 286(5439), 531–537. https://doi.org/10.1126/science.286.5439.531
Lundberg, S. M., & Lee, S.-I. (2017). A unified approach to interpreting model predictions. Advances in Neural Information Processing Systems, 30. https://proceedings.neurips.cc/paper_files/paper/2017/file/8a20a8621978632d76c43dfd28b67767-Paper.pdf
Zou, J., Huss, M., Abid, A., Mohammadi, P., Torkamani, A., & Telenti, A. (2019). A primer on deep learning in genomics. Nature Genetics, 51(1), 12–18. https://doi.org/10.1038/s41588-018-0295-5